


FDA: (De)Vice City
The obstacle is the opportunity when getting a product to market

Welcome to another edition of IF:Then, a weekly newsletter about legal strategy at the intersection of law and technology. To get this sweet content straight to your inbox each week, go ahead and subscribe below:

Regulatory strategy can be borne out of necessity: the need to quickly navigate a set of rules in order to get a business off the ground. It’s also borne out of opportunity: a path to operate and capitalize on or build a competitive advantage.
The FDA’s regulatory process for medical devices requires companies to balance both concepts. Given that the contours of modern FDA medical device regulation have been in place since the amendments to the Food, Drug, and Cosmetic Act in 1976, you might think there’s not a lot of wriggle room, but advancements in technology always present and opportunity to build a competitive advantage.
Below, we’ll talk about how cotton Q-tips, the Quip toothbrush, and a battle to measure your heart-rate reflect the considerations inherent in the FDA review process, and the delicate balance companies need to strike in order to leverage that process in a regulatory strategy.

Classification Station
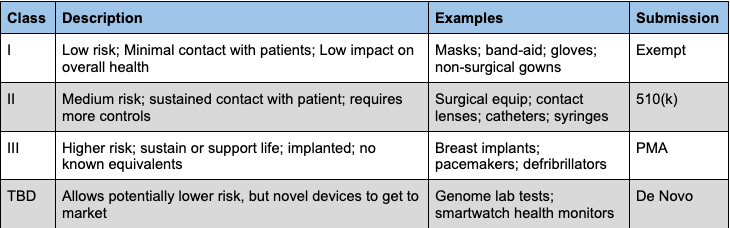
The definition of a “medical device” is broader than you’d think. It includes essentially any non-drug that “diagnoses, cures, mitigates, treats, or prevents a disease or condition” — so anything from band-aids and gloves to MRIs and CPAPs.
To get a new device to market, there are two related concepts a manufacturer needs to navigate: the classification and the regulatory pathway. Devices are slotted in Class I, II, or III, depending on their level of risk to the user, and the classification determines the regulatory submission pathway.
Premarket Notification (510(k))
Medical device regulatory pathways, or premarket submission types, set the standard of review by the FDA examiner. Class I devices are for the most part exempt from FDA reviews and can get to market quickly. No need for examiners to spend a ton of time evaluating the latest innovations from Sonicare if it’s just another powered toothbrush.

Class II devices for the most part require FDA “clearance,” obtained through a process called premarket notification, also referred to as 510(k). This type of application is centered around showing the device’s “substantial equivalence” to another device already on the market. Generally speaking, if the device’s features, intended use, and performance are similar to a “predicate device” that was already cleared, that is enough evidence for the FDA to deem the device as safe and effective.
")
Premarket Approval (PMA) and De Novo Review
Class III devices require a PMA submission. The idea around Class III is that life-altering devices require a high degree of scrutiny, as does anything that does not have substantial equivalence to a predicate device and is therefore, potentially innovative and new. This process almost certainly includes clinical trials, extensive data analysis, and many many conversations with your FDA examiner. Needless to say, this is probably an overly cumbersome process if you are introducing low-risk but novel device, like a new cotton swab that you’re actually supposed to use to clean your ears.

That is where the de novo pathway comes in handy. Introduced in 1997, a de novo submission allows a manufacturer to take advantage of a streamlined regulatory process where a low-risk device does not have a current classification. For the first couple decades this pathway was around, it was seldom used, but companies have started to think more strategically around FDA clearance, and the de novo application is proving to be a nice tool.
If you’d like to join the IF:Then Syndicate as an LP, or are in legal, compliance, or government affairs, and are interesting joining the IF:Then Community follow the link below:
FDA Review Meets Regulatory Strategy
On its face, the de novo pathway is an excellent tool to get a product to market, especially compare to the much more cumbersome PMA process which essentially assumes your product is high risk and goes from there. But de novo carries an inherent competitive disadvantage in a way, because it could allow a competitor to fast-follow with a 510(k) application using your device as the predicate. It’s almost like a reverse-patent, your work paves the road for your competitors. However, depending on how truly novel your underlying IP is, the de novo application does allow the sponsor to dictate the terms of clearance with respect to the capabilities and intent for use of the product. Pairing a de novo application with a strong patent can help you thread a regulatory needle by separating your novel product as opposed to expanding the follow-on capabilities of an existing product code like you would with a 510(k).
The Obstacle is the Opportunity

When you’re operating in a regulated space, seeing the obstacles in place as your opportunities is a difficult, but oftentimes rewarding way to view development. Many successful businesses have been built by making a hard thing easier (Square for small business merchants, Stripe for ecommerce), but sometimes being the only one willing or able to do the hard thing — or at least do it the fastest — is the way to a differentiated product. The more specific a de novo application is, the closer a follow-on competitor will have to come to your use case and performance. But of course the IP is the backstop — the closer they come, the more likely they are to infringe from a patent perspective.
AliveCor and Apple have spent the last few years doing the FDA/patent dance with their respective ECG devices tied to the Apple Watch. In 2012, AliveCor released its first FDA-cleared device “Kardia” — a smartphone case that doubled as an electrocardiogram (ECG) to measure and record electrical signals from the heart. In 2012 it released a credit card sized device. In 2015 the ECG readings went from physician-reviewed to analyzed by an algorithm, and in 2017 they released an AI model to learn to identify heart problems. That same year, they received clearance for the KardiaBand, an Apple Watch add-on providing ECG readings. Place your finger on the reader for 30 seconds, with results sent to the smartwatch.

AliveCor recently raised a $65 million Series E, and touts its FDA clearances in nearly all marketing materials.And why not? The company has successfully cleared 35 premarket notifications over the last 10 years — an impressive rate. They even received “breakthrough device designation” — which grants an accelerated approval process — for KardiaK, AliveCor’s AI-driven software platform that screened for hyperkalemia using ECG data rather than blood. What’s clear is that this company knew their way around the FDA clearance process, had optimized for speed and being first to market, and consistently iterated on its products along the way.
But while AliveCor might be masters of the 510(k), our overlords in Cupertino are truly the masters of rolling out an existing product and pretending it has never been done before. You come at the king, you best not miss.
In 2018, Apple received two de novo clearances on the same day for their own ECG and irregular heartbeat monitoring system. Why de novo when AliveCor had already receive a couple dozen 501(k)’s in the space? One reason must be that Apple simply didn’t care about speed, and that dictating the terms of the approval was far more important, because… well they’re Apple. And low and behold, a 2020 update allowed Apple to file a couple 510(k)s based on the substantial equivalence to its own devices — a much simpler process. Meanwhile, anyone else trying to follow not only needs to interact with Apple’s ecosystem, but they’d need to reach substantial equivalence based on the FDA labeling - the standards for features, performance, and use — that Apple itself directed. Tricky stuff.

Less than a year later AliveCor bailed on the KardiaBand entirely, pulling it from their store barely two years after FDA approval. Kudos to them for seeing the writing on the wall. Besides, they had new technology to worry about, going deeper into the segment with the KardiaMobile 6L, the “only personal 6-lead EKG in the world.” If you want a deep dive into the practical and medical differences between the KardiaMobile and the Apple Watch ECG capabilities, the “skeptical cardiologist” has you covered. But what I did find interesting was that despite ostensibly new technology, features, and use cases, AliveCor still went with the ‘ol reliable 510(k)! Additionally, the KardiaMobile 6L received clearance via substantial equivalence with both the previous Kardia offerings and the new category created by the Apple Watch.
More Like AliveCourt
Regulatory strategy is all fun and games, but eventually the litigators get involved. In late 2020 AliveCor sued Apple in up-and-coming-patent-litigation-haven Western District of Texas on three counts of patent infringement related to the ECGs. Earlier this year, AliveCor followed up with an antitrust suit in the Northern District of California. Here, AliveCor alleges that Apple essentially forced it to shut down the KardiaBand because Apple (1) owns the ecosystem these devices use to present results to the consumer; (2) began to copy Karida’s features; and (3) began giving AliveCor trouble with App Store Guidelines, constantly changing the rules or claiming violations and keeping AliveCor from maintaining accurate readings.
Whether most of that stuff is true or not, the way this has played out so far shines a light back on those FDA strategies. AliveCor is a three-part company — hardware, data analysis, and FDA expertise. They used that three-pronged approach to get products built, get them to market, and iterate on them. A de novo approach was probably not useful or necessary because they could patent their technology, and simply getting innovative technology to market would be their bread and butter. Apple, on the other hand, can leverage it owns the operating system as well as the hardware, and is extremely popular with its user base. Submitting de novo not only allowed them to dictate the follow-on terms, but laid the groundwork for showing differentiated technology from AliveCor in the inevitable suit. Meanwhile dozens of applications have been cleared by the FDA under the product code AliveCor built its business on, but only Apple, AliveCor, and Fitbit have released anything under the new code established by Apple. It’s probably not a regulatory moat, but it is an obstacle. Going forward, any device manufacturers in this space will need to grapple with their choice, and incorporate their decision into their clinical trials and/or data arguing to substantial equivalence.
It will be a while to see how these play out, and obviously this is just one example in a sea of years of FDA filings, but it’s interesting to see how things shake out. In any case, as we learn more, IF:Then will be there to take note.
Until next week, friends - David Ikenna Adams
Twitter | LinkedIn | Email | ifthen.vc